---
title: "Week 6 Lab"
author:
name: Alex Kaizer
roles: "Instructor"
affiliation: University of Colorado-Anschutz Medical Campus
toc: true
toc_float: true
toc-location: left
format:
html:
code-fold: show
code-overflow: wrap
code-tools: true
---
```{r, echo=F, message=F, warning=F}
library(kableExtra)
library(dplyr)
```
This page is part of the University of Colorado-Anschutz Medical Campus' [BIOS 6618 Labs](/labs/index.qmd) collection, the mini-lecture content delivered at the start of class before breaking out into small groups to work on the homework assignment.
# What's on the docket this week?
In Week 6 we are focusing on additional examples of bootstraps and permutation tests. Another helpful visual explainer of permutation tests is [about being an alpaca shepherd](https://www.jwilber.me/permutationtest/).
# A Colorful Illustration of Bootstraps and Permutations
There are a lot of similarities in between bootstraps and permutations:
* both involve resampling (with replacement for bootstraps, without replacement for permutations)
* both provide a means for evaluating the potential significance of a statistic (interpreting the confidence interval for bootstraps, interpreting a p-value for permutations)
* both can be thought of as nonparametric approaches (i.e., we don't have to assume any underlying distribution to conduct a test)
However, the nuances of sampling and what our ultimate goal for conducting either approach make them inherently different methods. If we strongly desire a p-value, we would implement a permutation test. Likewise, if we really wanted an estimate of the variability around a given statistic (e.g., its standard error or confidence interval), we would implement a bootstrap.
These concepts can be challenging to "picture" when considering data cases. As a change of pace, let's consider the "average" color of a given sample.
## The Colorful Dataset
Let's assume we have conducted a study with 6 observations in two groups, each represented by a color. Our "outcome" is the average color within the group (we can calculate this using the `average_colors()` function from the `miscHelpers` package that can be downloaded from GitHub):
```{r, class.source = 'fold-hide', message=F}
#| code-fold: true
# Run this function to install the kableExtra package to create some tables
#devtools::install_github("haozhu233/kableExtra")
library(kableExtra)
# Run this function to install the miscHelpers package to use the average_colors() function
#remotes::install_github("BenaroyaResearch/miscHelpers")
library(miscHelpers)
# Create vectors to store colors in
grp1 <- c('#0072B2','#0072B2','#0072B2','#97C9E4','#97C9E4','#97C9E4') # vector with 2 blues
grp2 <- c('#F0E442','#F0E442','#F0E442','#EA1F2F','#EA1F2F','#EA1F2F') # vector with reds and yellows
# Create matrix for 1:6 (group 1) and A:F (group 2)
grp_mat <- matrix( c(1:6,'Avg. 1','A','B','C','D','E','F','Avg. 2'), nrow=7, byrow=F, dimnames=list(c(paste0('Observation ',1:6),'Average'), c('Group 1','Group 2')))
grp_mat %>%
kbl(align='cc') %>%
kable_paper(full_width = F) %>%
column_spec(2, color = "black",
background = c(grp1, average_colors(grp1))) %>%
column_spec(3, color = "black",
background = c(grp2, average_colors(grp2))) %>%
row_spec(6, extra_css = "border-bottom: 2px solid")
```
We can see in the above that the average color in Group 1 is a blue that is the "average" of the 3 light and 3 dark blues. For Group 2 the average of 3 yellows and 3 reds is orange.
Let's first see an example of bootstrap resampling. Remember, here we sample *within* each group and *with replacement*:
```{r, class.source = 'fold-hide'}
#| code-fold: true
set.seed(515)
grp1b <- sort(sample(1:6, size=6, replace=T))
grp2b <- sort(sample(1:6, size=6, replace=T))
grp_matb <- matrix( c((1:6)[grp1b],'Avg. Boot 1',c('A','B','C','D','E','F')[grp2b],'Avg. Boot 2'), nrow=7, byrow=F)
colnames(grp_matb) <- c('Boot Group 1','Boot Group 2')
grp_combo <- cbind(grp_mat, '', grp_matb)
grp_combo %>%
kbl(align='cc') %>%
kable_paper(full_width = F) %>%
column_spec(2, color = "black",
background = c(grp1, average_colors(grp1))) %>%
column_spec(3, color = "black",
background = c(grp2, average_colors(grp2))) %>%
column_spec(5, color = "black",
background = c(grp1[grp1b], average_colors(grp1[grp1b]))) %>%
column_spec(6, color = "black",
background = c(grp2[grp2b], average_colors(grp2[grp2b]))) %>%
row_spec(6, extra_css = "border-bottom: 2px solid")
```
* In this bootstrap, we see that Boot Group 1 has resampled the 4th observation twice, so the 5th "light blue" observation is not included in the sample. However, since both observation 4 and 5 are "light blue" the overall average color is unchanged!
* In this bootstrap, we see that Boot Group 2 has resampled each "A", "B", and "C" twice...leaving no red observations! In this case the bootstrap distribution is only yellow observations (one potential extreme) and our average is simply yellow.
**How does this differ from a permutation test?** For the permutation test we combine all 12 observations before resampling *without replacement* as to who belongs to which group:
```{r, class.source = 'fold-hide'}
#| code-fold: true
set.seed(1012)
grp_perm <- sort(sample(1:12, size=6, replace=F)) # sample 6 observations to go into group 1, the rest will go into group 2
grp1p <- c(1:6,'A','B','C','D','E','F')[grp_perm] # take values according to index sampled for grp_perm
grp2p <- c(1:6,'A','B','C','D','E','F')[-grp_perm] # remove values according to index sampled from grp_perm
grp1p_col <- c(grp1,grp2)[grp_perm]
grp2p_col <- c(grp1,grp2)[-grp_perm]
grp_matp <- matrix( c(grp1p,'Avg. Perm 1',grp2p,'Avg. Perm 2'), nrow=7, byrow=F)
colnames(grp_matp) <- c('Perm Group 1','Perm Group 2')
grp_combo2 <- cbind(grp_combo, '', grp_matp)
grp_combo2 %>%
kbl(align='cc') %>%
kable_paper(full_width = F) %>%
column_spec(2, color = "black",
background = c(grp1, average_colors(grp1))) %>%
column_spec(3, color = "black",
background = c(grp2, average_colors(grp2))) %>%
column_spec(5, color = "black",
background = c(grp1[grp1b], average_colors(grp1[grp1b]))) %>%
column_spec(6, color = "black",
background = c(grp2[grp2b], average_colors(grp2[grp2b]))) %>%
column_spec(8, color = "black",
background = c(grp1p_col, average_colors(grp1p_col))) %>%
column_spec(9, color = "black",
background = c(grp2p_col, average_colors(grp2p_col))) %>%
row_spec(6, extra_css = "border-bottom: 2px solid")
```
* In our permutation sample we see that group membership has been broken, so that members of the original Group 1 and Group 2 are now part of the Perm Group 1 and Perm Group 2. This process helps us to break any potential association that may have existed previously (e.g., only blues in one group versus red and yellows in another).
* We see our average colors are now an interesting brownish (Avg. Perm 1) and grayish-blue (Avg. Perm 2).
* If our original observation (e.g., the difference in sample means between groups) was actually from the *null distribution*, then we would expect that estimate to fall near the center of our permutation distribution. In our color example, the original colors are decided in our two camps (blue vs. red/yellow), so here we see a muddier picture of the average color.
Based on either approach, we would want to conduct a large number of bootstrap or permutation resamples, and then examine the overall distribution:
```{r, class.source = 'fold-hide'}
#| code-fold: true
bp_mat <- matrix('', ncol=5,nrow=100)
colnames(bp_mat) <- c('Boot Group 1','Boot Group 2','','Perm Group 1','Perm Group 2')
rownames(bp_mat) <- paste0('Simulation ',1:100)
for(j in 1:100){
set.seed(2020+j)
# permutation resample
grp_perm <- sample(1:12, size=6, replace=F) # sample 6 observations to go into group 1, the rest will go into group 2
bp_mat[j,'Perm Group 1'] <- average_colors(c(grp1,grp2)[grp_perm])
bp_mat[j,'Perm Group 2'] <- average_colors(c(grp1,grp2)[-grp_perm])
# bootstrap resample
bp_mat[j,'Boot Group 1'] <- average_colors( grp1[ sample(1:6,size=6,replace=T) ])
bp_mat[j,'Boot Group 2'] <- average_colors( grp2[ sample(1:6,size=6,replace=T) ])
}
# Object to create kable from
bp_mat_kableshell <- matrix('', ncol=5,nrow=100)
colnames(bp_mat_kableshell) <- c('Boot Group 1','Boot Group 2','','Perm Group 1','Perm Group 2')
rownames(bp_mat_kableshell) <- paste0('Simulation ',1:100)
bp_mat_kableshell %>%
kbl() %>%
kable_paper(full_width = F) %>%
column_spec(2, color = "black",
background = bp_mat[,'Boot Group 1']) %>%
column_spec(3, color = "black",
background = bp_mat[,'Boot Group 2']) %>%
column_spec(5, color = "black",
background = bp_mat[,'Perm Group 1']) %>%
column_spec(6, color = "black",
background = bp_mat[,'Perm Group 2'])
```
If this were an actual study with a numeric outcome we could describe the variability of our average color within or between groups (**bootstrap sampling**) or we could calculate if our observed data is more extreme than the null/permutation distribution (**permutation test**).
Indeed we can see that for all 100 bootstrap samples, there are various shades of blue for Group 1 and red/orange/yellow for Group 2. Whereas for all 100 permutation samples there is a range of colors from purpleish to blueish to orangeish to greenish...a random combination of our colors!
# Bootstrap and Permutation Test Example: The Ratio of Sample Variances
Assume we have conducted a randomized trial that enrolled 100 total participants with celiac disease to examine the potential effect of a new vaccine ($T$) to desensitize the immune system in its reaction to gluten as compared to a placebo ($P$). The outcome is the measure of the tissue transglutaminase IgA antibody (tTG-IgA).
As a secondary analysis of the primary trial, we wish to explore if the variability of tTG-IgA in the two groups with 50 participants each are approximately equal, even if their mean tTG-IgA concentrations are different. To evaluate this, we propose using the ratio of the placebo to the treatment group:
\[ \frac{s_{P}^2}{s_{T}^2} \]
If the ratio is equal to 1, our estimates are the same (i.e., our null hypothesis). Let's assume that we have zero idea what distribution this may take, so we want to explore using bootstraps to describe the variability or a permutation test to calculate a p-value summary if our sample is unexpected.
## Null Scenario
Let's start with the null scenario, where both groups have the same variance. We will simulate from:
\[ Y_{P} \sim \text{Gamma}(\text{shape}=10, \text{scale}=3), \; Y_{T} \sim \text{Gamma}(\text{shape}=5, \text{scale}=\sqrt{18}) \]
These parameters were chosen so that both sets of data will have a variance of 90 (i.e., the variance for the gamma distribution when parameterized with the shape ($k$) and scale ($\theta$) is $k\theta^2$).
```{r, class.source = 'fold-show'}
set.seed(612)
placebo <- rgamma(n=50, shape=10, scale=3)
vaccine <- rgamma(n=50, shape=5, scale=sqrt(18))
var(placebo) # calculate the sample variances
var(vaccine) # calculate the sample variances
obs_ratio <- var(placebo)/var(vaccine) # calculate the ratio of the variances
obs_ratio
```
## **Null Scenario: Bootstrap**
Let's conduct a bootstrap with 10,000 resamples of our ratio of the sample variances to describe the variability of this statistic:
```{r, class.source = 'fold-show'}
B <- 10^4 #set number of bootstraps
var_ratio <- numeric(B) #initialize vector to store results in
nP <- length(placebo) #sample size of placebo group
nT <- length(vaccine) #sample size of vaccine group
set.seed(612) #set seed for reproducibility
for (i in 1:B){
placebo.boot <- sample(placebo, nP, replace=T)
vaccine.boot <- sample(vaccine, nT, replace=T)
var_ratio[i] <- var(placebo.boot) / var(vaccine.boot)
}
```
Let's now visualize the shape of our bootstrap distribution:
```{r, class.source = 'fold-show'}
par(mfrow=c(1,2)) #create plotting area for 2 figures in one row
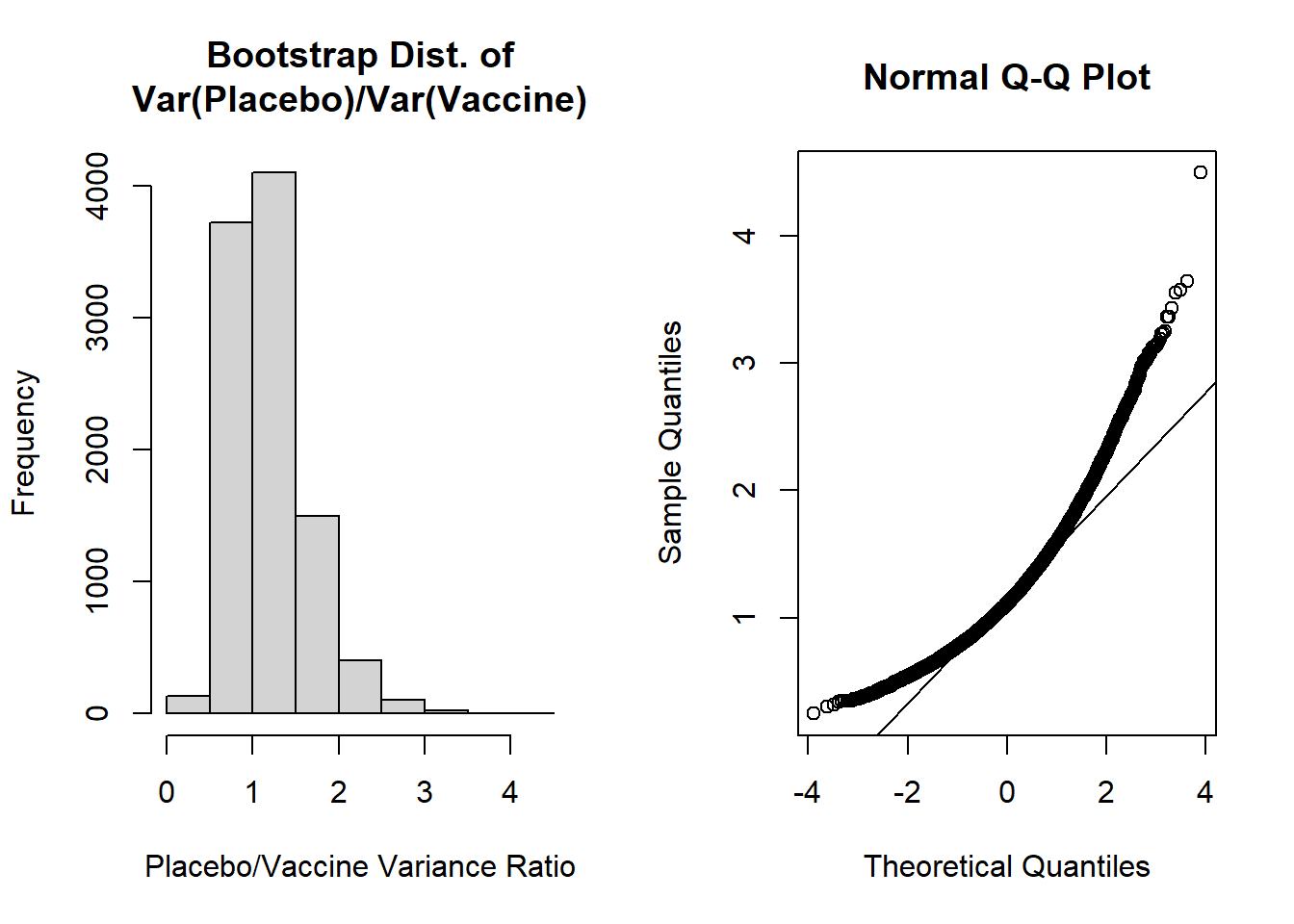
hist(var_ratio, main='Bootstrap Dist. of\nVar(Placebo)/Var(Vaccine)', xlab='Placebo/Vaccine Variance Ratio')
qqnorm(var_ratio); qqline(var_ratio)
```
The shapes of these plots suggest the ratio of variances is not normally distributed. Our histogram is right skewed and the normal Q-Q plot deviates from the diagonal line.
Let's then calculate the mean, SE, and bias of the bootstrap distribution:
```{r, class.source = 'fold-show'}
mean(var_ratio) # bootstrap mean ratio
mean(var_ratio)-obs_ratio # bias for ratio
sd(var_ratio) # bootstrap SE
(mean(var_ratio)-obs_ratio) / sd(var_ratio) # estimate of accuracy
```
The most concerning aspect of this is the bias/SE estimate > 0.10, suggesting our bootstrap percentile intervals may not be very accurate. However, let's calculate the 95% bootstrap percentile interval as our "best" approach given the two options from our lecture (i.e., normal percentile or bootstrap percentile):
```{r, class.source = 'fold-show'}
quantile( var_ratio, c(0.025,0.975))
```
For the 95% bootstrap percentile CI, we are 95% confident that the true ratio of variances falls between 0.546 and 2.269. Additionally, because it is estimated from our data directly, 95% of the bootstrap estimates of ratios of variances fall in this interval.
The accuracy of our bootstrap percentile can be estimated by the ratio of the bias/SE, which we noted was 0.215. Since this exceeds +0.10 we may be concerned about the accuracy of this estimate.
It could also be noted that our 95% bootstrap percentile CI includes 1, so we would fail to reject the null hypothesis.
## **Null Scenario: Permutation Test**
Perhaps we are more interested in calculating a p-value to determine if the sample ratio differs from its underlying null distribution:
```{r, class.source = 'fold-show'}
B <- 10^4 - 1 #set number of times to complete permutation sampling
result <- numeric(B)
nP <- length(placebo)
obs_ratio <- var(placebo)/var(vaccine) # calculate the ratio of the variances
set.seed(612) #set seed for reproducibility
pool_dat <- c(placebo, vaccine)
for(j in 1:B){
index <- sample(x=1:length(pool_dat), size=nP, replace=F)
placebo_permute <- pool_dat[index]
vaccine_permute <- pool_dat[-index]
result[j] <- var(placebo_permute) / var(vaccine_permute)
}
# Histogram
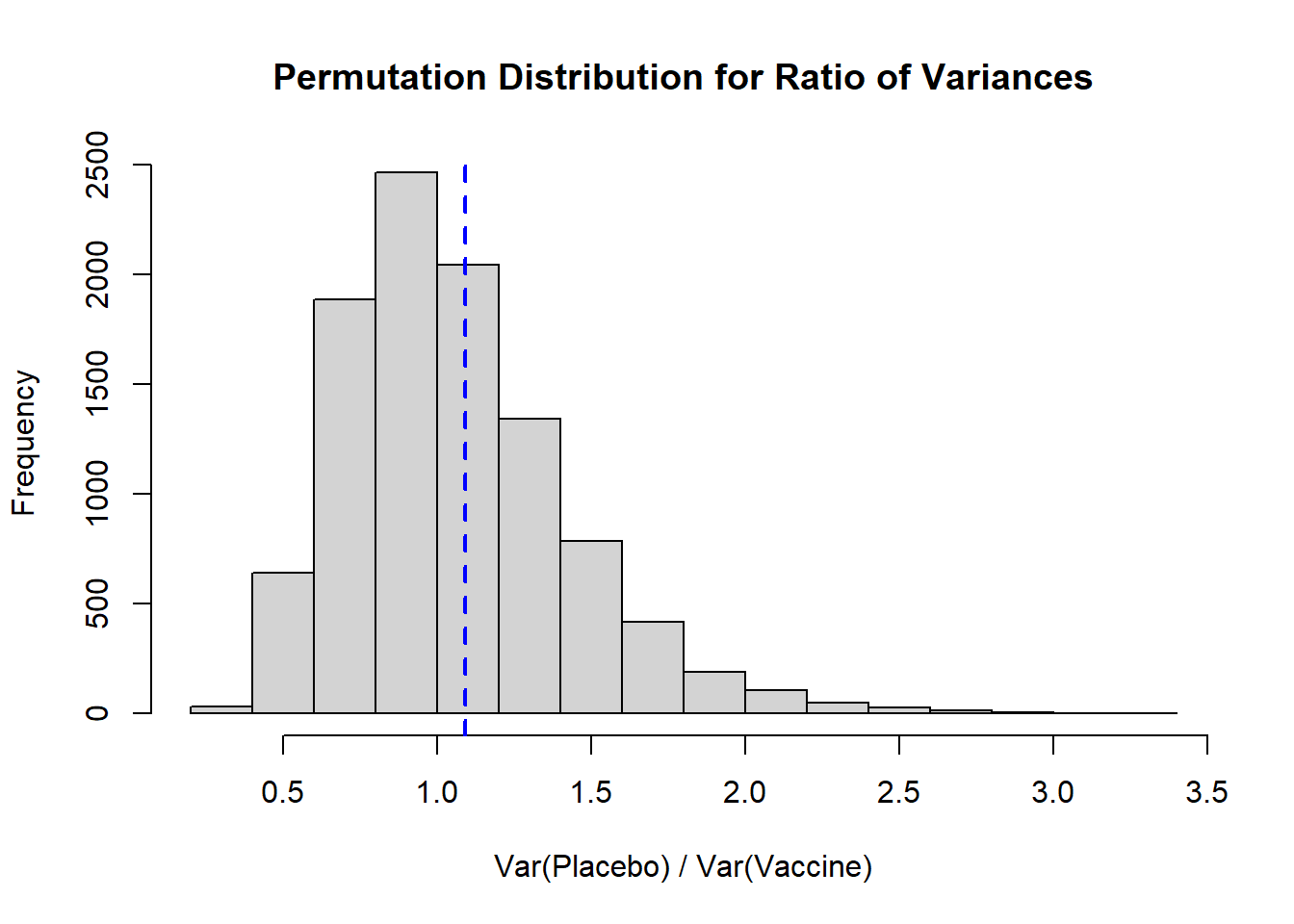
hist( result, xlab='Var(Placebo) / Var(Vaccine)',
main='Permutation Distribution for Ratio of Variances')
abline( v=obs_ratio, lty=2, col='blue', lwd=2)
```
Again, we see a distribution that is right skewed, and our observed ratio of variances falls fairly close to our expected null of 1. To calculate a **two-sided p-value** we would take the larger of the proportion of our distribution that falls above our observed ratio or, in our context, the proportion that falls below `1/obs_ratio` (the inverse of our observed ratio), and multiply it by 2:
```{r, class.source = 'fold-show'}
#note, we take the larger p-value and multiply by 2 (as compared to replacing <= with >)
((sum(result >= obs_ratio) + 1)/(B+1))
((sum(result <= (1/obs_ratio)) + 1)/(B+1))
# Calculate permutation p-value for two-sided test
2 * ((sum(result <= (1/obs_ratio)) + 1)/(B+1))
```
Here we see that our two-sided p-value is 0.8018.
A few important notes here:
* It is helpful to plot the permutation distribution to note what direction ($\leq$ vs. $\geq$) we need to use in our calculation.
* We multiple the larger p-value by 2 to (1) account for the two-sided test and (2) to be more conservative (vs. using the smaller proportion).
* If we wanted to calculate a **one-sided p-value** we would need to define that null and alternative hypothesis. For example, if our $H_0$ is that the ratio of variances is larger than 1 (i.e., the placebo group has larger variance), we would specifically interpret our result as $p=0.3992$. If the null hypothesis is that the ratio of variance is smaller than 1, based on our observed ratio we would have $1-0.3992=0.6008$.
## Alternative Scenario
Let's check an alternative scenario, where we will simulate from:
\[ Y_{P} \sim \text{Gamma}(\text{shape}=10, \text{scale}=3), \; Y_{T} \sim \text{Gamma}(\text{shape}=5, \text{scale}=3) \]
These parameters were chosen so that the placebo has a true variance of 90 and the vaccine has a true variance of 45 (i.e., the variance for the gamma distribution when parameterized with the shape ($k$) and scale ($\theta$) is $k\theta^2$).
```{r, class.source = 'fold-show'}
set.seed(312)
placebo <- rgamma(n=50, shape=10, scale=3)
vaccine <- rgamma(n=50, shape=5, scale=3)
var(placebo) # calculate the sample variances
var(vaccine) # calculate the sample variances
obs_ratio <- var(placebo)/var(vaccine) # calculate the ratio of the variances
obs_ratio
```
## **Alternative Scenario: Bootstrap**
Let's conduct a bootstrap with 10,000 resamples of our ratio of the sample variances to describe the variability of this statistic:
```{r, class.source = 'fold-show'}
B <- 10^4 #set number of bootstraps
var_ratio <- numeric(B) #initialize vector to store results in
nP <- length(placebo) #sample size of placebo group
nT <- length(vaccine) #sample size of vaccine group
set.seed(312) #set seed for reproducibility
for (i in 1:B){
placebo.boot <- sample(placebo, nP, replace=T)
vaccine.boot <- sample(vaccine, nT, replace=T)
var_ratio[i] <- var(placebo.boot) / var(vaccine.boot)
}
```
Let's now visualize the shape of our bootstrap distribution:
```{r, class.source = 'fold-show'}
par(mfrow=c(1,2)) #create plotting area for 2 figures in one row
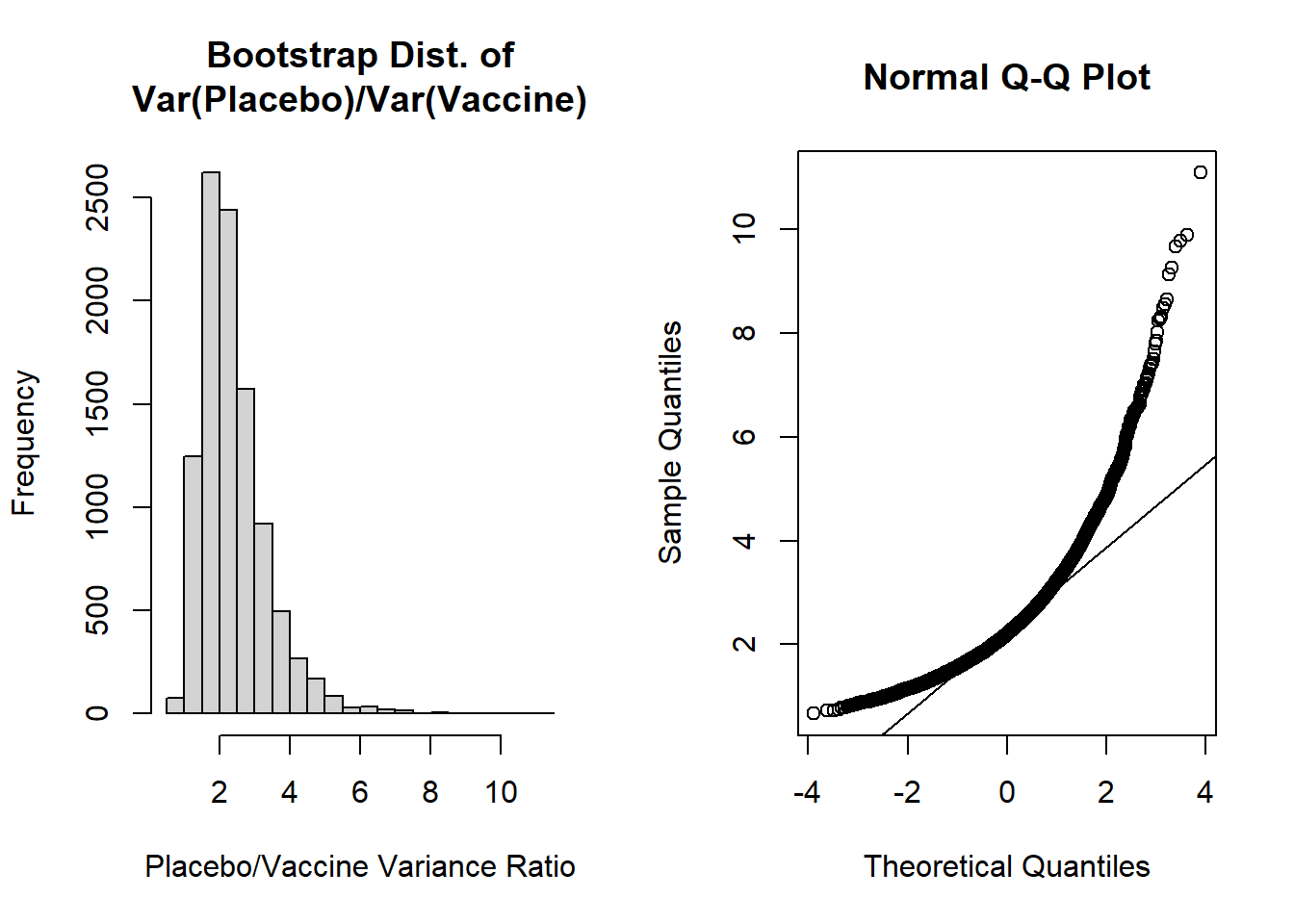
hist(var_ratio, main='Bootstrap Dist. of\nVar(Placebo)/Var(Vaccine)', xlab='Placebo/Vaccine Variance Ratio')
qqnorm(var_ratio); qqline(var_ratio)
```
The shapes of these plots suggest the ratio of variances is not normally distributed. Our histogram is right skewed and the normal Q-Q plot deviates from the diagonal line.
Let's then calculate the mean, SE, and bias of the bootstrap distribution:
```{r, class.source = 'fold-show'}
mean(var_ratio) # bootstrap mean ratio
mean(var_ratio)-obs_ratio # bias for ratio
sd(var_ratio) # bootstrap SE
(mean(var_ratio)-obs_ratio) / sd(var_ratio) # estimate of accuracy
```
The most concerning aspect of this is the bias/SE estimate > 0.10, suggesting our bootstrap percentile intervals may not be very accurate. However, let's calculate the 95% bootstrap percentile interval as our "best" approach given the two options from our lecture (i.e., normal percentile or bootstrap percentile):
```{r, class.source = 'fold-show'}
quantile( var_ratio, c(0.025,0.975))
```
For the 95% bootstrap percentile CI, we are 95% confident that the true ratio of variances falls between 1.151 and 4.787. Additionally, because it is estimated from our data directly, 95% of the bootstrap estimates of ratios of variances fall in this interval.
The accuracy of our bootstrap percentile can be estimated by the ratio of the bias/SE, which we noted was 0.243. Since this exceeds +0.10 we may be concerned about the accuracy of this estimate.
It could also be noted that our 95% bootstrap percentile CI excludes 1, so we may conclude that we would reject the null hypothesis, and conclude our ratio of sample variances are not equal. Further, given the ratio as placebo/vaccine, we could conclude that the placebo has greater variability.
## **Alternative Scenario: Permutation Test**
Perhaps we are more interested in calculating a p-value to determine if the sample ratio differs from its underlying null distribution:
```{r, class.source = 'fold-show'}
B <- 10^4 - 1 #set number of times to complete permutation sampling
result <- numeric(B)
nP <- length(placebo)
obs_ratio <- var(placebo)/var(vaccine) # calculate the ratio of the variances
set.seed(312) #set seed for reproducibility
pool_dat <- c(placebo, vaccine)
for(j in 1:B){
index <- sample(x=1:length(pool_dat), size=nP, replace=F)
placebo_permute <- pool_dat[index]
vaccine_permute <- pool_dat[-index]
result[j] <- var(placebo_permute) / var(vaccine_permute)
}
# Histogram
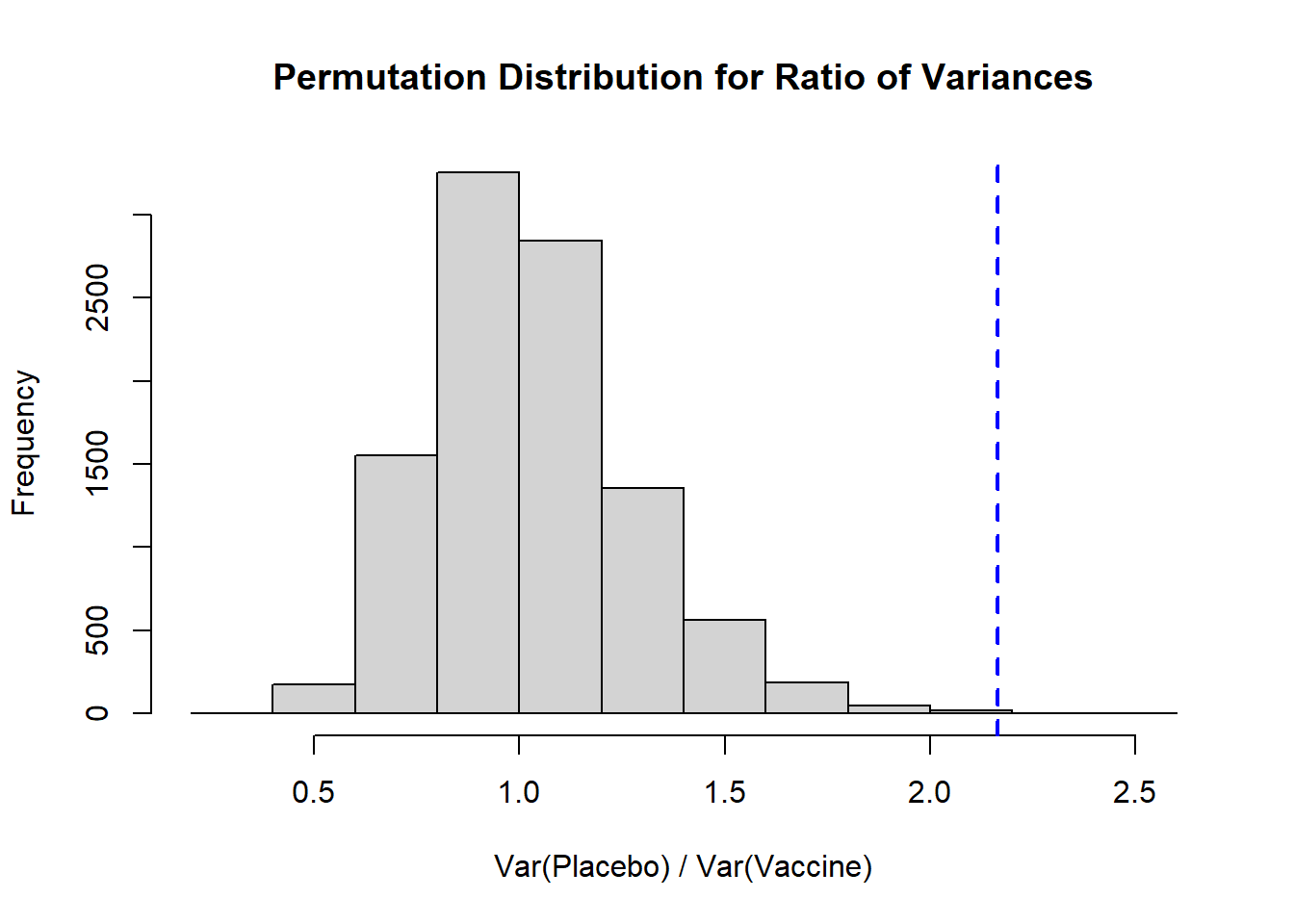
hist( result, xlab='Var(Placebo) / Var(Vaccine)',
main='Permutation Distribution for Ratio of Variances')
abline( v=obs_ratio, lty=2, col='blue', lwd=2)
```
Again, we see a distribution that is right skewed, and our observed ratio of variances falls fairly close to our expected null of 1. To calculate a **two-sided p-value** we would take the larger of the proportion of our distribution that falls above our observed ratio or, in our context, the proportion that falls below `1/obs_ratio` (the inverse of our observed ratio), and multiply it by 2:
```{r, class.source = 'fold-show'}
#note, we take the larger p-value and multiply by 2 (as compared to replacing <= with >)
((sum(result >= obs_ratio) + 1)/(B+1))
((sum(result <= (1/obs_ratio)) + 1)/(B+1))
# Calculate permutation p-value for two-sided test
2 * ((sum(result <= (1/obs_ratio)) + 1)/(B+1))
```
Here we see that our two-sided p-value is 0.0016, so we would reject the null hypothesis that the variances are equal for placebo and vaccine groups.
A few important notes here:
* It is helpful to plot the permutation distribution to note what direction ($\leq$ vs. $\geq$) we need to use in our calculation.
* If we wanted to calculate a **one-sided p-value** we would need to define that null and alternative hypothesis. For example, if our $H_0$ is that the placebo has a larger variance than the vaccine group, we would conclude that the placebo group does appear to have a larger variance based on our one-sided $p=0.0006$.
* If the null hypothesis is that the vaccine has a larger variance than the placebo (i.e., the ratio is <1), based on our observed ratio we would have $p=1-0.0006=0.9994$, or we would fail to reject that null hypothesis. In other words, we cannot conclude that the vaccine group has a larger variance than the placebo.
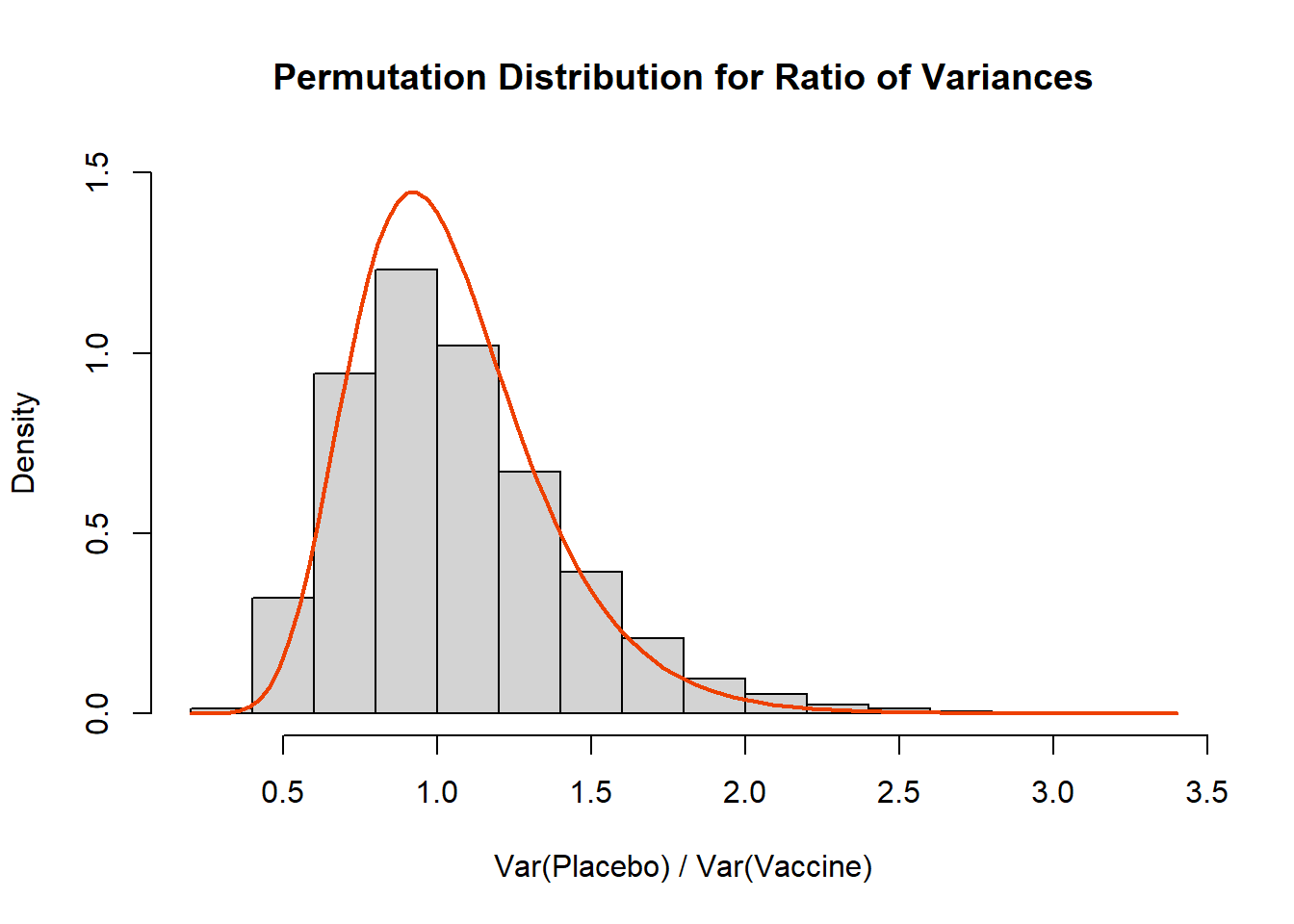
## Wait a Minute, What is the Distribution of the Ratio of Variances??
Ahh, we almost snuck away without addressing the theoretical distribution! Generally speaking the ratio of variances will follow an $F_{n_1-1,n_2-1}$ distribution under the null hypothesis that the variances are equal:
```{r, class.source = 'fold-hide'}
#| code-fold: true
set.seed(612)
placebo <- rgamma(n=50, shape=10, scale=3)
vaccine <- rgamma(n=50, shape=5, scale=sqrt(18))
B <- 10^4 - 1 #set number of times to complete permutation sampling
result <- numeric(B)
nP <- length(placebo)
obs_ratio <- var(placebo)/var(vaccine) # calculate the ratio of the variances
set.seed(612) #set seed for reproducibility
pool_dat <- c(placebo, vaccine)
for(j in 1:B){
index <- sample(x=1:length(pool_dat), size=nP, replace=F)
placebo_permute <- pool_dat[index]
vaccine_permute <- pool_dat[-index]
result[j] <- var(placebo_permute) / var(vaccine_permute)
}
# Histogram
hist( result, xlab='Var(Placebo) / Var(Vaccine)',
main='Permutation Distribution for Ratio of Variances', freq=F, ylim=c(0,1.5))
curve(df(x,df1=49,df2=49),add=T, lwd=2, col='orangered2')
```